Tìm hiểu về rối loạn chuyển hóa axit amin
Rối loạn chuyển hoá axit amin nguy hiểm như thế nào?
Khi cơ thể chúng ta ăn thức ăn có đạm (protein), protein sẽ được chuyển hoá thành axit amin (9 axit amin cần thiết và 12 axit amin không cần thiết). Các axit amin sẽ tiếp tục được chuyển hoá để cung cấp năng lượng và cấu tạo nên cơ thể.
Khi bị thiếu hụt một trong các enzyme chuyển hoá axit amin thì con đường chuyển hoá sẽ bị tắc nghẽn. Kết quả dẫn tới các chất chuyển hoá trước chỗ tắc tăng quá mức bình thường và gây độc cho các tế bào và các cơ quan trong cơ thể. Các cơ quan bị gây độc nhiều nhất là não, gan, thận, tim.
Rối loạn chuyển hóa axit amin là hệ tiêu hóa không chuyển hóa được thức ăn thành dưỡng chất
Ví dụ: Trong bệnh Mapple Syrup Urine Disease (bệnh nước tiểu mùi siro cây phong) là thiếu các enzyme chuyển hoá dẫn tới tăng quá mức 3 axit amin cần thiết Leucine, Isoleucine, Valine. Kết quả là gây tổn thương não, gan do tăng Leucine quá mức. Hay trong bệnh rối loạn chuyển hoá propionic máu là thiếu các enzyme chuyển hoá propionyl – CoA dẫn đến tăng cao lượng axit propionic trong máu làm tổn thương các tế bào não và tuỷ xương.
Nguyên nhân
Đột biến các gen chịu trách nhiệm sản xuất các enzyme chuyển hoá axit amin.
Biểu hiện lâm sàng
Bệnh có thể gặp ở mọi lứa tuổi nhưng thường biểu hiện bệnh trước 18 tháng tuổi. Biểu hiện triệu chứng lâm sàng đa dạng, không điển hình nên dễ bị chẩn đoán nhầm với các bệnh lý viêm não – màng não, nhiễm trùng huyết, bại não. Các biểu hiện lâm sàng điển hình của rối loạn chuyển hoá axit amin:
- Các triệu chứng thần kinh cấp tính: hôn mê, thất điều, hội chứng não cấp
- Các đợt bệnh kéo dài hoặc thoái triển mà không rõ tình trạng nhiễm trùng đặc hiệu.
- Các triệu chứng thần kinh tiến triển: sa sút trí tuệ, giảm thị lực, thính lực…
- Các biểu hiện bệnh lý ở đa cơ quan.
Chẩn đoán xác định
Dựa vào các xét nghiệm hoá sinh hiện đại như định lượng axit amin, phân tích axit amin hữu cơ niệu và phân tích gen gây bệnh.
Nhóm bệnh này có thể chẩn đoán sớm trước khi xuất hiện triệu chứng lâm sàng qua chương trình sàng lọc sơ sinh các nhóm bệnh rối loạn chuyển hoá.
Điều trị
- Chế độ ăn: Hạn chế đạm (protein) 1 – 1,2g/kg/ngày, tuỳ theo lứa tuổi, tuỳ từng bệnh nhân và tuỳ từng thời kỳ của bệnh. Tăng cường lượng tinh bột và dầu/mỡ, các vitamin và yếu tố vi lượng cần thiết hàng ngày.
- Các thuốc tăng cường chuyển hoá và đào thải chất độc: L carnitine, Biotin, Vitamin B12, Vitamin B1, Vitamin B12, Arginnine, Natribenzoat,…
- Các đợt cấp: Nhập viện, theo dõi, truyền glucose liều cao, truyền lipit, lọc máu,…
Theo dõi
Thăm khám định kỳ, kiểm tra các chức năng sinh hoá và huyết học, phát triển thể chất và tinh thần…
Tư vấn di truyền
Quy luật di truyền lặn trên nhiễm sắc thể thường
- Bệnh có tính chất ngắt quãng qua các thế hệ.
- Khả năng mắc bệnh ở nam và nữ như nhau.
- Khả năng di truyền cho các thế hệ sau:
Nếu bố, mẹ là người mang gen thì khả năng 25% số con bình thường, 25% số con có biểu hiện bệnh và 50% số con là người mang gen giống bố mẹ.
Nếu chỉ có bố hoặc mẹ là người mang gen thì thế hệ sau không có con bị biểu hiện bệnh, 50% số con là người mang gen, 50% số con hoàn toàn bình thường.
Sàng lọc sơ sinh được thực hiện cho trẻ từ 36 – 72 giờ đầu sau sinh bằng cách lấy máu gót chân, giúp phát hiện sớm nhiều nhóm bệnh khi chưa có triệu chứng:
55 bệnh rối loạn bẩm sinh axid amin, acid hữu cơ, acid béo
Suy giáp bẩm sinh
Tăng sản thượng thận bẩm sinh
Thiếu men G6PD
Các bệnh dự trữ thể tiêu bào: Pompe, Gaucher, MPS1, Fabry, Niemann Pick, Krabbe…
Các rối loạn tổng hợp Hemonglobin
Suy giảm miễn dịch bẩm sinh thể phối hợp và nặng
Bệnh Galactosemia
Theo Bệnh viện Nhi Trung ương
 Từ khóa:
Từ khóa:

- Sản phẩm vì sức khỏe
-
Đau họng, ợ hơi, tức ngực: Người đàn ông ngỡ ngàng phát hiện ung thư đại tràng sớm qua nội soi
Người đàn ông 37 tuổi khỏe mạnh bàng hoàng phát hiện có tổn thương nguy cơ ung thư nhờ công nghệ nội soi hiện đại tại Bệnh viện Thu Cúc. Câu chuyện của anh là hồi chuông cảnh tỉnh cho nhiều người trẻ còn đang chủ quan, thờ ơ với sức khỏe tiêu hóa.July 2 at 10:13 am
-
Lấy lại nụ cười tự tin với dịch vụ bọc răng sứ tại TCI
Hàng nghìn khách hàng đã tìm lại nụ cười tự tin nhờ dịch vụ bọc răng sứ tại Khoa Răng Hàm Mặt bệnh viện Đa khoa Quốc tế (ĐKQT) Thu Cúc TCI.July 1 at 12:01 pm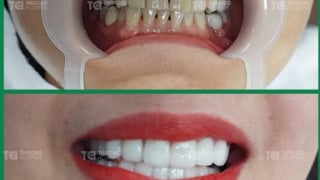
-
Chấm dứt ợ hơi, ợ chua, mất ngủ nhờ phương pháp đo áp lực thực quản HRM hiện đại
Từng khổ sở với ợ hơi, ợ chua, nuốt vướng và mất ngủ triền miên, chị H.T.B.L (38 tuổi) đã tìm lại giấc ngủ ngon và sức khỏe chỉ sau thời gian ngắn điều trị tại Bệnh viện Đa khoa Quốc tế Thu Cúc nhờ kỹ thuật đo áp lực và nhu động thực quản độ phân giải cao HRM.July 1 at 12:00 pm
-
Long Châu hợp tác cùng Viện Kiểm nghiệm ATVSTP Quốc gia chủ động tiên phong tiến hành “kiểm tra kép vì sức khỏe người dân và khách hàng”
Trước thực trạng thuốc giả, thực phẩm chức năng giả làm ảnh hưởng nghiêm trọng đến niềm tin của người tiêu dùng và sức khỏe cộng đồng, hưởng ứng chủ trương của Chính phủ trong phòng chống hàng giả, hàng kém chất lượng, hệ thống nhà thuốc Long Châu chủ động tiên phong ký kết hợp tác chiến lược với Viện Kiểm nghiệm an toàn vệ sinh thực phẩm Quốc gia (Bộ Y tế).June 25 at 5:09 pm